
Introduction to Single Cell RNA-Seq Part 3: Normalize and scale
Set up workspace
First, we need to load the required libraries.
library(Seurat)
library(kableExtra)
library(ggplot2)
library(dplyr)
We will be continuing the work from Part 1 and so need to load in the RDS file
experiment.aggregate <- readRDS("scRNA_workshop-02.rds")
experiment.aggregate
## An object of class Seurat
## 38606 features across 40052 samples within 1 assay
## Active assay: RNA (38606 features, 0 variable features)
## 1 layer present: counts
Lets go ahead and set that common seed for everyone
set.seed(12345)
Normalize the data
After filtering, the next step is to normalize the data. We employ a global-scaling normalization method, LogNormalize, that normalizes the gene expression measurements for each cell by the total expression, multiplies this by a scale factor (10,000 by default), and then log-transforms the data.
?NormalizeData
experiment.aggregate <- NormalizeData(
object = experiment.aggregate,
normalization.method = "LogNormalize",
scale.factor = 10000)
experiment.aggregate
## An object of class Seurat
## 38606 features across 40052 samples within 1 assay
## Active assay: RNA (38606 features, 0 variable features)
## 2 layers present: counts, data
The function produces a new layer in the object called “data”. We can now access the normalised data using LayerData. We can use this to show that we can get a list of the most highly expressed genes overall.
norm.expression <- LayerData(experiment.aggregate,layer="data")
mean.expression <- apply(norm.expression, 1, mean)
names(mean.expression) <- rownames(experiment.aggregate)
mean.expression <- sort(mean.expression, decreasing = TRUE)
head(mean.expression, n=50)
## FTH1 MT-CO1 FTL CD74 B2M TMSB4X MALAT1 MT-CO2
## 4.646231 4.539242 4.525198 4.489474 4.458964 4.284744 4.198105 3.860866
## ACTB EEF1A1 HLA-DRA TPT1 MT-CO3 HLA-B VIM HLA-A
## 3.855886 3.711285 3.653941 3.587522 3.534209 3.503641 3.407929 3.285413
## HLA-DRB1 TMSB10 S100A11 RPLP1 MT-CYB IFI30 HLA-C RPL28
## 3.276857 3.240642 3.203091 3.174771 3.137407 3.085733 3.068303 3.030152
## RPL41 RPL10 PFN1 MT-ND4L MT-ATP6 TYROBP HLA-DPA1 MT-ND4
## 2.953827 2.952570 2.910644 2.886496 2.880725 2.875505 2.870495 2.859882
## S100A6 MT-ND5 MYL6 RPL13A MT-ND2 RPS8 RPL13 PSAP
## 2.846236 2.844457 2.785106 2.785023 2.764556 2.721428 2.716863 2.712893
## RPS19 MT-ND1 LYZ RPS12 CST3 CTSD RPS2 RPLP2
## 2.709430 2.688671 2.657034 2.656320 2.655618 2.648870 2.647233 2.607146
## HLA-DPB1 HLA-DRB5
## 2.599841 2.597642
We see a number of Mitochondrial genes that are highly expressed as well as some of our Ribosomal products.
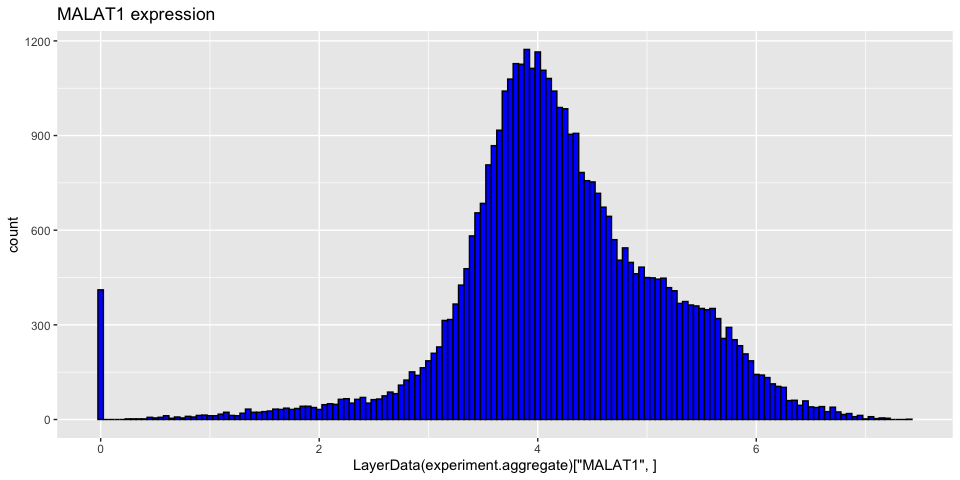 Explore a couple more of these genes
Explore a couple more of these genes
Cell cycle assignment
Cell cycle phase can be a significant source of variation in single cell and single nucleus experiments. There are a number of automated cell cycle stage detection methods available for single cell data. For this workshop, we will be using the built-in Seurat cell cycle function, CellCycleScoring. This tool compares gene expression in each cell to a list of cell cycle marker genes and scores each barcode based on marker expression. The phase with the highest score is selected for each barcode. Seurat includes a list of cell cycle genes in human single cell data.
Seurat comes with the human gene lists needed for Cell Cycle determination.
cc.genes.updated.2019
## $s.genes
## [1] "MCM5" "PCNA" "TYMS" "FEN1" "MCM7" "MCM4"
## [7] "RRM1" "UNG" "GINS2" "MCM6" "CDCA7" "DTL"
## [13] "PRIM1" "UHRF1" "CENPU" "HELLS" "RFC2" "POLR1B"
## [19] "NASP" "RAD51AP1" "GMNN" "WDR76" "SLBP" "CCNE2"
## [25] "UBR7" "POLD3" "MSH2" "ATAD2" "RAD51" "RRM2"
## [31] "CDC45" "CDC6" "EXO1" "TIPIN" "DSCC1" "BLM"
## [37] "CASP8AP2" "USP1" "CLSPN" "POLA1" "CHAF1B" "MRPL36"
## [43] "E2F8"
##
## $g2m.genes
## [1] "HMGB2" "CDK1" "NUSAP1" "UBE2C" "BIRC5" "TPX2" "TOP2A"
## [8] "NDC80" "CKS2" "NUF2" "CKS1B" "MKI67" "TMPO" "CENPF"
## [15] "TACC3" "PIMREG" "SMC4" "CCNB2" "CKAP2L" "CKAP2" "AURKB"
## [22] "BUB1" "KIF11" "ANP32E" "TUBB4B" "GTSE1" "KIF20B" "HJURP"
## [29] "CDCA3" "JPT1" "CDC20" "TTK" "CDC25C" "KIF2C" "RANGAP1"
## [36] "NCAPD2" "DLGAP5" "CDCA2" "CDCA8" "ECT2" "KIF23" "HMMR"
## [43] "AURKA" "PSRC1" "ANLN" "LBR" "CKAP5" "CENPE" "CTCF"
## [50] "NEK2" "G2E3" "GAS2L3" "CBX5" "CENPA"
For other species, a user-provided gene list may be substituted, or the orthologs of the human gene list used instead. Do not run the code below for human experiments!
# mouse code DO NOT RUN for human data
library(biomaRt)
convertHumanGeneList <- function(x){
require("biomaRt")
human = useEnsembl("ensembl",
dataset = "hsapiens_gene_ensembl",
mirror = "uswest")
mouse = useEnsembl("ensembl",
dataset = "mmusculus_gene_ensembl",
mirror = "uswest")
genes = getLDS(attributes = c("hgnc_symbol"),
filters = "hgnc_symbol",
values = x ,
mart = human,
attributesL = c("mgi_symbol"),
martL = mouse,
uniqueRows=T)
humanx = unique(genes[, 2])
print(head(humanx)) # print first 6 genes found to the screen
return(humanx)
}
# convert lists to mouse orthologs
s.genes <- convertHumanGeneList(cc.genes.updated.2019$s.genes)
g2m.genes <- convertHumanGeneList(cc.genes.updated.2019$g2m.genes)
Once an appropriate gene list has been identified, the CellCycleScoring function can be run.
experiment.aggregate <- CellCycleScoring(experiment.aggregate,
s.features = cc.genes.updated.2019$s.genes,
g2m.features = cc.genes.updated.2019$g2m.genes,
set.ident = TRUE)
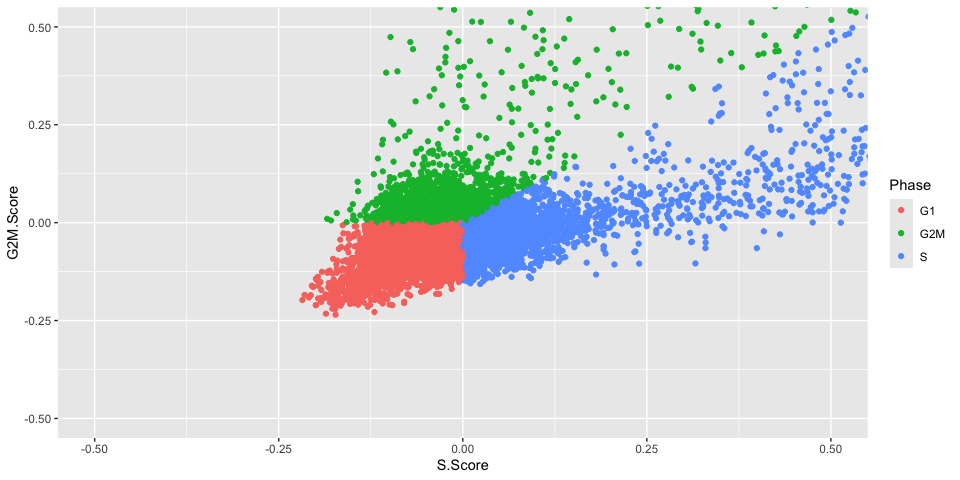
table(experiment.aggregate[["Phase"]]) %>%
kable(caption = "Number of Cells in each Cell Cycle Stage",
col.names = c("Stage", "Count"),
align = "c") %>%
kable_styling()
| Stage | Count |
|---|---|
| G1 | 24873 |
| G2M | 4501 |
| S | 10678 |
We can visualize how the algorithm assigns state based on the computed S.Score and G2M.Score
Because the “set.ident” argument was set to TRUE (this is also the default behavior), the active identity of the Seurat object was changed to the phase. To return the active identity to the sample identity, use the Idents function.
table(Idents(experiment.aggregate))
##
## G1 S G2M
## 24873 10678 4501
Idents(experiment.aggregate) <- "orig.ident"
table(Idents(experiment.aggregate))
##
## LRTI_WRK1 LRTI_WRK2 LRTI_WRK3 LRTI_WRK4
## 12372 12480 2084 13116
Identify variable genes
The function FindVariableFeatures identifies the most highly variable genes (default 2000 genes) by fitting a line to the relationship of log(variance) and log(mean) using loess smoothing, uses this information to standardize the data, then calculates the variance of the standardized data. This helps avoid selecting genes that only appear variable due to their expression level.
?FindVariableFeatures
experiment.aggregate <- FindVariableFeatures(
object = experiment.aggregate,
selection.method = "vst") ## vst having issues??
length(VariableFeatures(experiment.aggregate))
## [1] 2000
The function places data in an object called HVFInfo, we can extract and look at the results of find variable features for each gene.
## mean variance variance.expected variance.standardized
## SPP1 3.1527514 2686.06256 13.3812444 70.68009
## WFDC2 2.3125936 1828.29703 8.0168136 70.56587
## SERPINB3 0.9510137 328.98015 2.0923136 68.68482
## SLPI 1.8165635 1393.96303 5.4465838 64.16529
## BPIFB1 2.5344552 3206.84176 9.3129384 61.54649
## CCL17 1.7835314 377.07781 5.2919254 60.07977
## TPSAB1 0.4262459 112.10639 0.7843354 60.06400
## PTGDS 0.4777789 98.86088 0.8900317 58.90872
## CPA3 0.3148157 56.25267 0.5730767 57.78090
## LCN2 1.0441676 712.20201 2.3810364 57.29496
## CCL20 1.0298362 136.89489 2.3354899 56.27489
## RIMS1 0.6606162 87.75036 1.3035991 54.89394
## CCL2 2.0587486 499.79263 6.6439926 50.74984
## SCGB3A1 2.6130031 6191.75650 9.7929258 50.11783
## PIGR 0.5649905 78.83029 1.0793266 49.02960
## KRT19 0.6037152 81.37270 1.1681873 47.55942
## CXCL10 3.1525267 624.45735 13.3796467 46.31380
## MSMB 0.9065215 886.44963 1.9604775 43.78314
## PPBP 0.3517677 81.13333 0.6405779 43.03506
## SCGB1A1 3.1295316 13863.77504 13.2166073 40.59522
We can plot this data, while marking the top 20
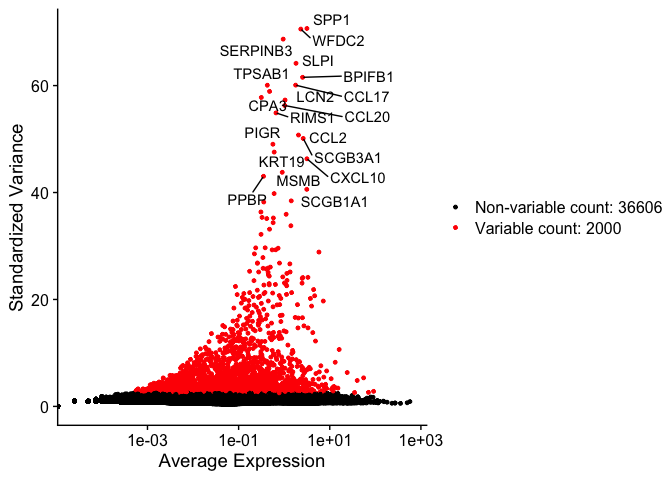
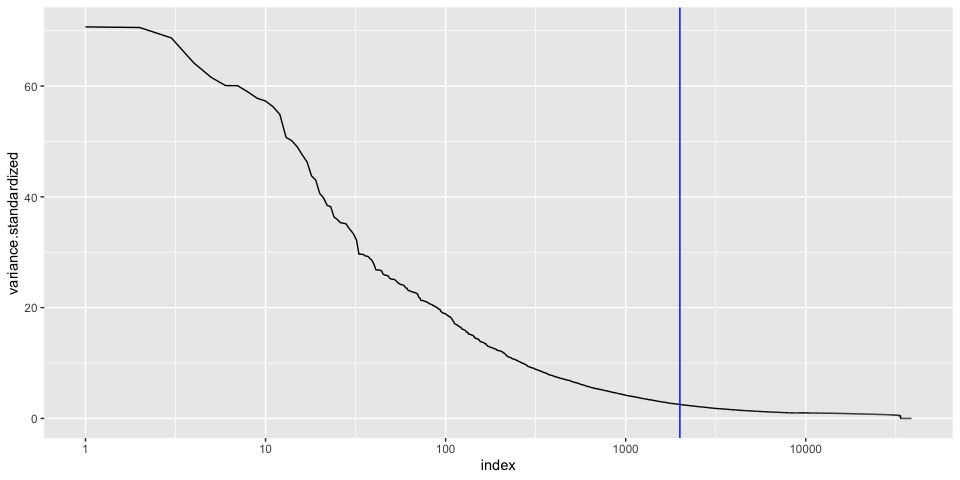
Goes mostly flat at around 100, so we will keep our list of variable genes
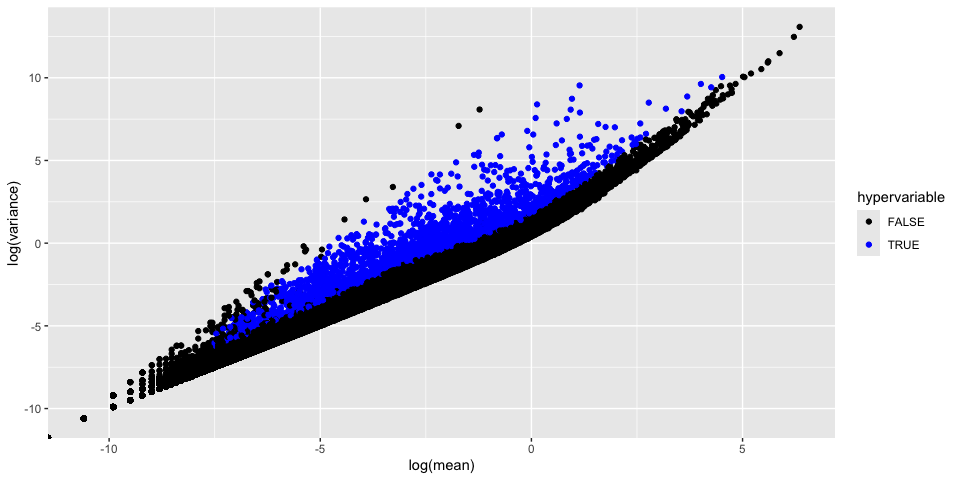
How do the results change if you use selection.method = “dispersion” or selection.method = “mean.var.plot”?
FindVariableFeatures isn’t the only way to set the “variable features” of a Seurat object. Another reasonable approach is to select a set of “minimally expressed” genes.
min.value <- 2
min.cells <- 10
num.cells <- Matrix::rowSums(GetAssayData(experiment.aggregate, slot = "count") > min.value)
genes.use <- names(num.cells[which(num.cells >= min.cells)])
length(genes.use)
## [1] 15841
VariableFeatures(experiment.aggregate) <- genes.use
Prepare for the next section
Save object
saveRDS(experiment.aggregate, file = "scRNA_workshop-03.rds")
Download Rmd
download.file("https://raw.githubusercontent.com/ucsf-cat-bioinformatics/2024-08-SCRNA-Seq-Analysis/main/data_analysis/04-dimensionality_reduction.Rmd", "04-dimensionality_reduction.Rmd")
Session Information
sessionInfo()
## R version 4.4.1 (2024-06-14)
## Platform: aarch64-apple-darwin20
## Running under: macOS Sonoma 14.6.1
##
## Matrix products: default
## BLAS: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRblas.0.dylib
## LAPACK: /Library/Frameworks/R.framework/Versions/4.4-arm64/Resources/lib/libRlapack.dylib; LAPACK version 3.12.0
##
## locale:
## [1] en_US.UTF-8/en_US.UTF-8/en_US.UTF-8/C/en_US.UTF-8/en_US.UTF-8
##
## time zone: America/Los_Angeles
## tzcode source: internal
##
## attached base packages:
## [1] stats graphics grDevices utils datasets methods base
##
## other attached packages:
## [1] dplyr_1.1.4 ggplot2_3.5.1 kableExtra_1.4.0 Seurat_5.1.0
## [5] SeuratObject_5.0.2 sp_2.1-4
##
## loaded via a namespace (and not attached):
## [1] deldir_2.0-4 pbapply_1.7-2 gridExtra_2.3
## [4] rlang_1.1.4 magrittr_2.0.3 RcppAnnoy_0.0.22
## [7] spatstat.geom_3.3-2 matrixStats_1.3.0 ggridges_0.5.6
## [10] compiler_4.4.1 systemfonts_1.1.0 png_0.1-8
## [13] vctrs_0.6.5 reshape2_1.4.4 stringr_1.5.1
## [16] pkgconfig_2.0.3 fastmap_1.2.0 labeling_0.4.3
## [19] utf8_1.2.4 promises_1.3.0 rmarkdown_2.28
## [22] purrr_1.0.2 xfun_0.47 cachem_1.1.0
## [25] jsonlite_1.8.8 goftest_1.2-3 highr_0.11
## [28] later_1.3.2 spatstat.utils_3.1-0 irlba_2.3.5.1
## [31] parallel_4.4.1 cluster_2.1.6 R6_2.5.1
## [34] ica_1.0-3 spatstat.data_3.1-2 bslib_0.8.0
## [37] stringi_1.8.4 RColorBrewer_1.1-3 reticulate_1.38.0
## [40] spatstat.univar_3.0-0 parallelly_1.38.0 lmtest_0.9-40
## [43] jquerylib_0.1.4 scattermore_1.2 Rcpp_1.0.13
## [46] knitr_1.48 tensor_1.5 future.apply_1.11.2
## [49] zoo_1.8-12 sctransform_0.4.1 httpuv_1.6.15
## [52] Matrix_1.7-0 splines_4.4.1 igraph_2.0.3
## [55] tidyselect_1.2.1 abind_1.4-5 rstudioapi_0.16.0
## [58] yaml_2.3.10 spatstat.random_3.3-1 codetools_0.2-20
## [61] miniUI_0.1.1.1 spatstat.explore_3.3-2 listenv_0.9.1
## [64] lattice_0.22-6 tibble_3.2.1 plyr_1.8.9
## [67] withr_3.0.1 shiny_1.9.1 ROCR_1.0-11
## [70] evaluate_0.24.0 Rtsne_0.17 future_1.34.0
## [73] fastDummies_1.7.4 survival_3.7-0 polyclip_1.10-7
## [76] xml2_1.3.6 fitdistrplus_1.2-1 pillar_1.9.0
## [79] KernSmooth_2.23-24 plotly_4.10.4 generics_0.1.3
## [82] RcppHNSW_0.6.0 munsell_0.5.1 scales_1.3.0
## [85] globals_0.16.3 xtable_1.8-4 glue_1.7.0
## [88] lazyeval_0.2.2 tools_4.4.1 data.table_1.15.4
## [91] RSpectra_0.16-2 RANN_2.6.2 leiden_0.4.3.1
## [94] dotCall64_1.1-1 cowplot_1.1.3 grid_4.4.1
## [97] tidyr_1.3.1 colorspace_2.1-1 nlme_3.1-166
## [100] patchwork_1.2.0 cli_3.6.3 spatstat.sparse_3.1-0
## [103] spam_2.10-0 fansi_1.0.6 viridisLite_0.4.2
## [106] svglite_2.1.3 uwot_0.2.2 gtable_0.3.5
## [109] sass_0.4.9 digest_0.6.37 progressr_0.14.0
## [112] ggrepel_0.9.5 farver_2.1.2 htmlwidgets_1.6.4
## [115] htmltools_0.5.8.1 lifecycle_1.0.4 httr_1.4.7
## [118] mime_0.12 MASS_7.3-61